{kind=link}

كان أسامة في الشهر السادس من عمره عندما بدأت أعراض مرضيّة مثيرة للقلق في الظهور عليه. حمى مرتفعة وآلام شديدة تجعله لا يكف عن البكاء طوال اليوم، وكان يمسك برأسه وقدميه دائمًا وهو يتلوى من الوجع.
لم يجد الأطباء تفسيرًا لما يحدث بعد أن استبعدوا احتمالًا تلو الآخر، ومع مرور الوقت ازدادت الأعراض سوءًا فانتفخت بطنه، وتورمت أصابع يديه وقدميه، وبات يقضي ليله بين الأنين والسّهاد.
استمرت الأعراض الغامضة ونوبات الألم التي تلمّ به لعام ونصف والأطباء في حيرة من أمرهم، حتى لاح دليل طبي فكَّ الغموض الذي اكتنف الحالة، إذ انتبه أحد الأطباء إلى اصفرار عينيّ الصغير ونتائج فحوصات دمه، التي أشارت إلى فقر دم حاد ونسبة عالية من مركب البيلوروبين، الناتج من تحلل كرات الدم الحمراء.
عُرض أسامة على طبيبة أمراض الدم بمستشفى طرابلس للأطفال، فطلبت المزيد من الفحوص وطرحت بعض الأسئلة عن تاريخ العائلة وصلة القرابة بين الزوجين، وبعد اطلاعها على النتائج أعلنت لوالدي أسامة التشخيص النهائي؛ ابنهما مصاب بالأنيميا المنجلية.
آلام شديدة
الأنيميا المنجلية واحدة من اضطرابات خلايا الدم الحمراء الموروثة، حيث يتغير شكل كريات الدم الحمراء المسؤولة عن نقل الأكسجين إلى كل أعضاء وأنسجة الجسم، فالكريات الطبيعية تكون دائرية الشكل مقعرة الجانبين، أما في حالة الطفل أسامة فهي هلالية أو منجلية الشكل، ومن هنا جاءت تسمية هذا المرض.
كرية الدم الحمراء الحاملة للمطفرة المنجلية (يسار) مقارنة بكرية دم حمراء طبيعية (يمين)
تجد خلايا أسامة الحمراء صعوبة في المرور من الأوعية الدموية، كما أنها لا تنقل الأكسجين بشكل كافٍ إلى خلايا الجسم الأخرى، فعندما يتعرض للجفاف أو الإرهاق أو الأجواء شديدة البرودة أو الحرارة، فإن الأكسجين ينخفض بشكل أكبر، ويمر الطفل بنوبة من الآلام الشديدة.
فيما تعيش كريات الدم الطبيعية من 90 إلى 120 يومًا، فإن كريات الدم المنجلية لا تصمد سوى لعشرة إلى عشرين يومًا، ومن ثم تتحلل مسببة فقرًا في الدم أو ما اصطلح على تسميته بـالأنيميا [1].
مضاعفات خطيرة
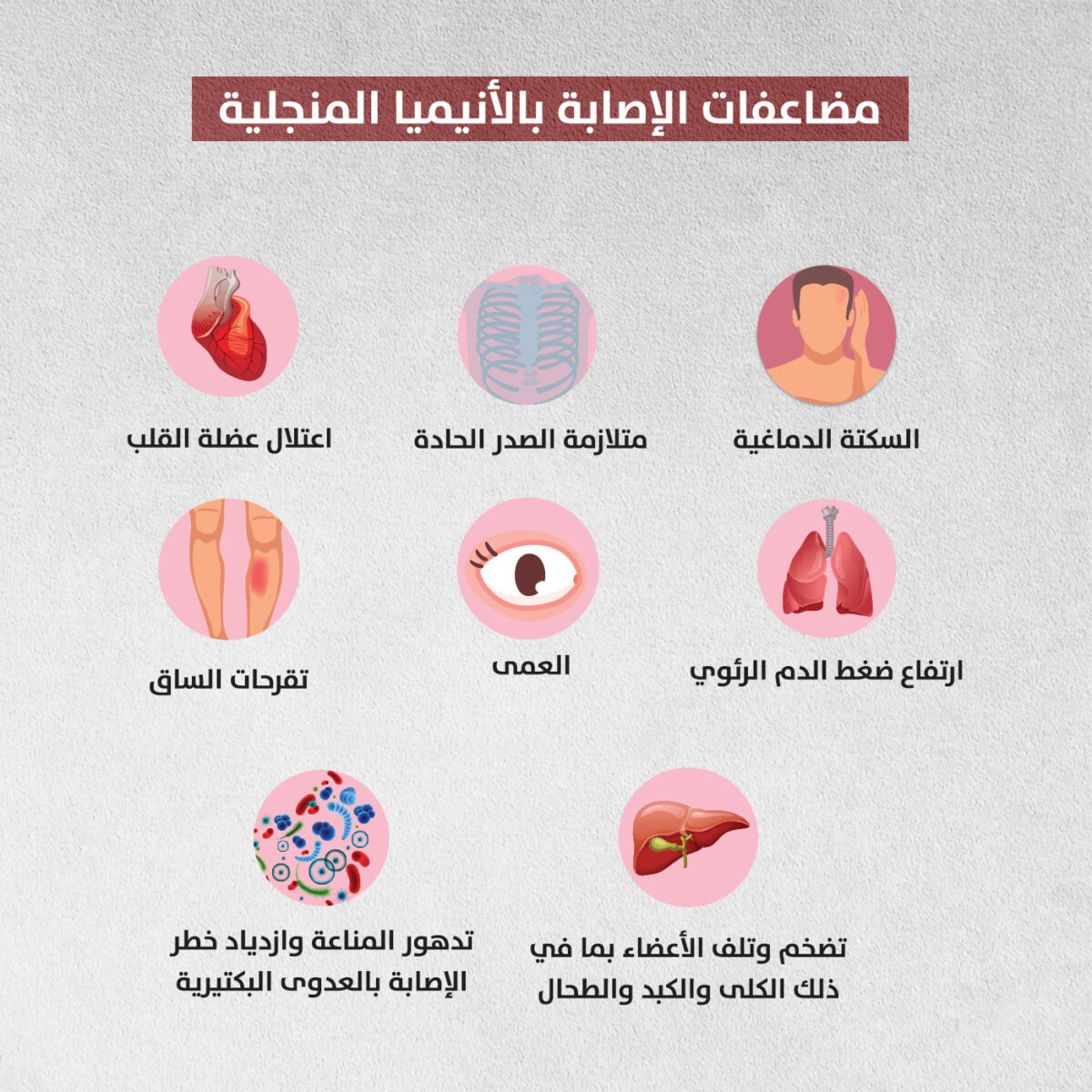
يؤدي الشكل المنجلي غير الطبيعي لخلايا الدم الحمراء إلى انحشارها داخل الأوعية الدموية، ما قد يتسبب في مضاعفات شديدة الخطورة كتلف الأعضاء والسكتات الدماغية والعمى [2].
في ديسمبر/ كانون الأول عام 2012 عندما كان أسامة في السادسة من عمره تدهورت حالته بشكل مفاجئ، مع نوبة جديدة من الألم صاحبها صداع رهيب وحرارة عالية بلغت 41 درجة مئوية، وأخذت الآلام منه كل مأخذ ولم تفلح معها كل أنواع المسكنات التي وصفها له الأطباء، ثم ارتفع ضغط دمه بشكل خطير، وأصيب بقصور في القلب والكلى حتى صار لون بوله بنيًا قاتمًا، ما استوجب إدخاله العناية الفائقة.
تبين أن أسامة يمر بإحدى مضاعفات الأنيميا المنجلية، وهو ما يعرف باحتباس الكبد (Hepatic sequestration)، حيث انحشرت خلايا الدم الحمراء داخل الكبد فتضخم واضطربت أنزيماته بشكل جنوني، ظل الصغير تحت المتابعة الدقيقة من الأطباء الذين نقلوا الدم إليه بشكل مستمر، وهو ما أنقذ حياته وأخرجه من العناية الفائقة بعد أسبوع حرج، مر على أسرته وكأنه دهر.
حرف واحد يفسد الهيموجلوبين
تُنتج الخلايا الجذعية في نخاع العظام كريات الدم الحمراء، وينتهي الأمر بالكريات الناضجة إلى مجرى الدم بعد أن تتخلص من نواتها، لتفسح المجال أمام 200 إلى 300 جزيء من الهيموجلوبين المسؤول عن الارتباط بالأكسجين [3].
يتركب جزيء الهيموجلوبين من أربعة سلاسل بروتينية، اثنتان تسميان بجلوبين – ألفا، واثنتان تسميان بجلوبين – بيتا، وسلاسل بيتا هي ما تهمنا الآن في فهمنا للأنيميا المنجلية.
تتكون سلسلة جلوبين – بيتا من 146 حمضًا أمينيًا (الأحماض الأمينية هي اللبنة الرئيسية في تركيب البروتينات) ترتيبها وأنواعها محفوظة مسبقًا في جيناتنا وتتصل ببعضها بشكل أشبه بحبات المسبحة.
في الأنيميا المنجلية يحدث خطأ في ترتيب هذه الأحماض بسبب طفرة في الجين المسؤول عن تصنيع بروتين جلوبين – بيتا، هذه الطفرة تتمثل في تغير لحرف واحد في شفرة الحمض الأميني السادس من GAG إلى GTG فيصير الحمض الأميني “الفالين” بديلًا عن “الجلوتامين”.
تغير هذا الحمض الأميني يجعل جزئيات الهيموجلوبين تضطرب وترتبط ببعضها البعض داخل كرية الدم الحمراء مكونة هياكل كبيرة صلبة تشبه الحبال، ما يمنح كرية الدم الحمراء شكلها المنجلي ويفقدها مرونتها التي كانت تتمتع بها [4].
دون أن يعلم
يمتلك كل إنسان 46 كورموسومًا (23 زوجًا) داخل نواة خلاياه، تحتوي كل المعلومات والصفات الوراثية، منها 22 زوجًا من الكروموزومات الجسدية وزوج واحد من الكروموزومات الجنسية التي تحدد جنسه.
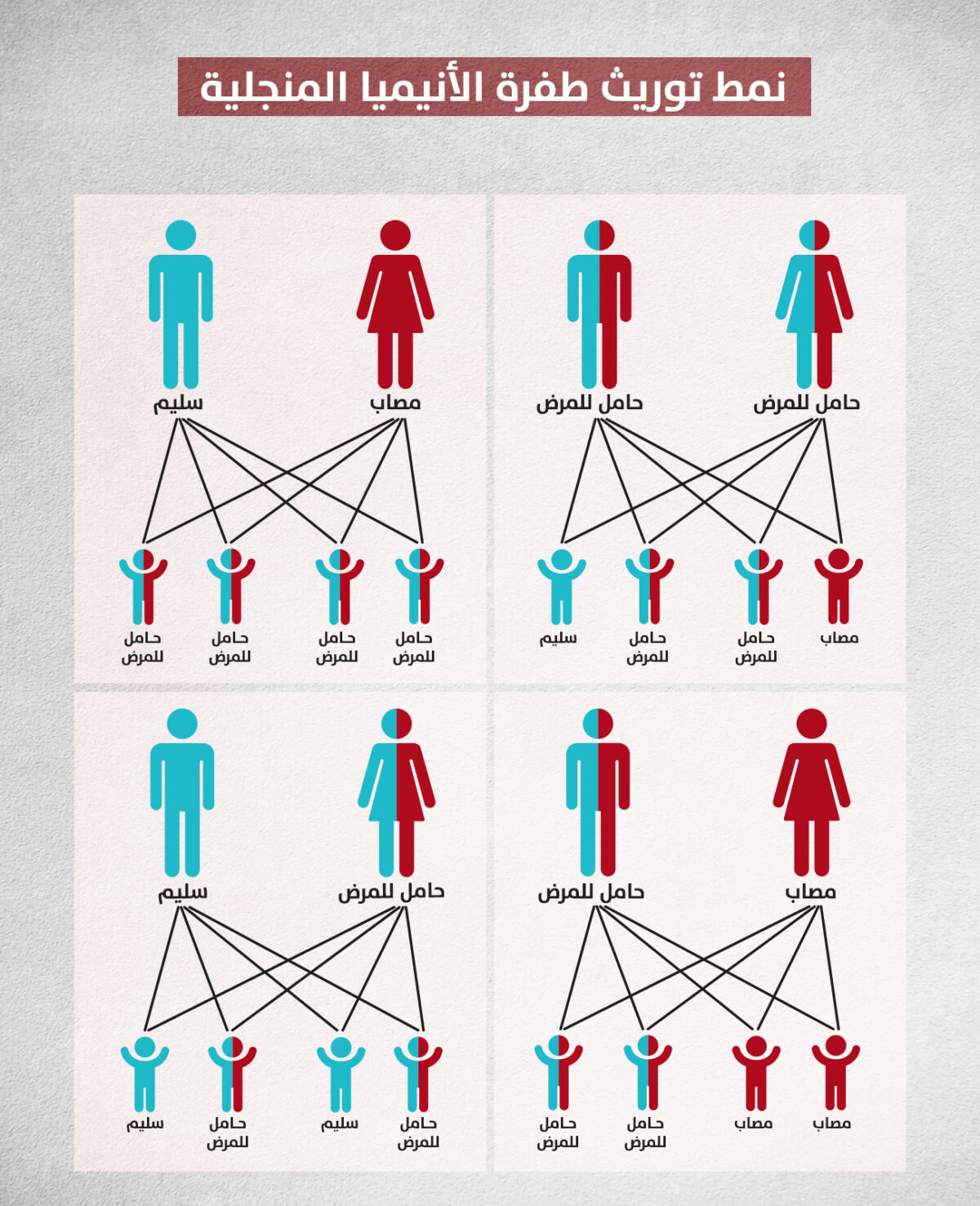
يقع جين الجلوبين – بيتا في زوج الكروموزوم الجسدي رقم 11 وبالتالي فإن كل مولود يرث نسختين من جين الجلوبين – بيتا، واحد من أبيه وآخر من أمه، فإذا كانت إحدى نسختي الجين تحتوي على طفرة الأنيميا المنجلية يعتبر المولود حاملًا للمرض ولا تظهر عليه أعراض، أما إذا كانت النسختان تحتويان على الطفرة يعتبر المولود مصابا بالأنيميا المنجلية، ويسمى هذا النمط بالوراثة المتنحية [1,3].
حالات توريث طفرة الأنيميا المنجلية بنمط الوراثة الجسدية المتنحية
{kind=link}
في حالة أسامة كان كلا والديه حاملًا للمرض، دون أن يعلم، وبهذا النمط الوراثي فإن 25% من أبنائهما سيكونون مصابين بالأنيميا المنجلية. تزداد احتمالية تلاقي أبوين حاملين للمرض في حالات زواج الأقارب، بسبب استمرار توريث الطفرة المتنحية للأنيميا المنجلية عبر الأجيال المتعاقبة.
تقول الدكتورة هويدا الحاجي، إنها شهدت عشرات الحالات من الأنيميا المنجلية، وجلها لصغار تربط آباءهم صلة قربى.
“الصدمة ثم الإحساس بالذنب مع مرور الزمن، هذا حال كل أبوين حاملين للمرض بعدما أخبرهما بنتائج تحليل الهيموجلوبين”، تتابع الحاجي.
مدن الجنوب الأكثر تأثرًا
في الواقع لا توجد إحصائيات أو أرقام رسمية من الدولة الليبية عن نسبة انتشار الأنيميا المنجلية، لكن بعض الشواهد والدراسات العلمية تشير إلى ارتفاع نسبة هذا المرض خاصة في الجنوب الليبي.
كما تؤكد الحاجي أن أغلب المصابين بالأنيميا المنجلية المترددين إليها يأتون من مدن الجنوب، ومن تاورغاء شرق طرابلس، والعجيلات غربها.
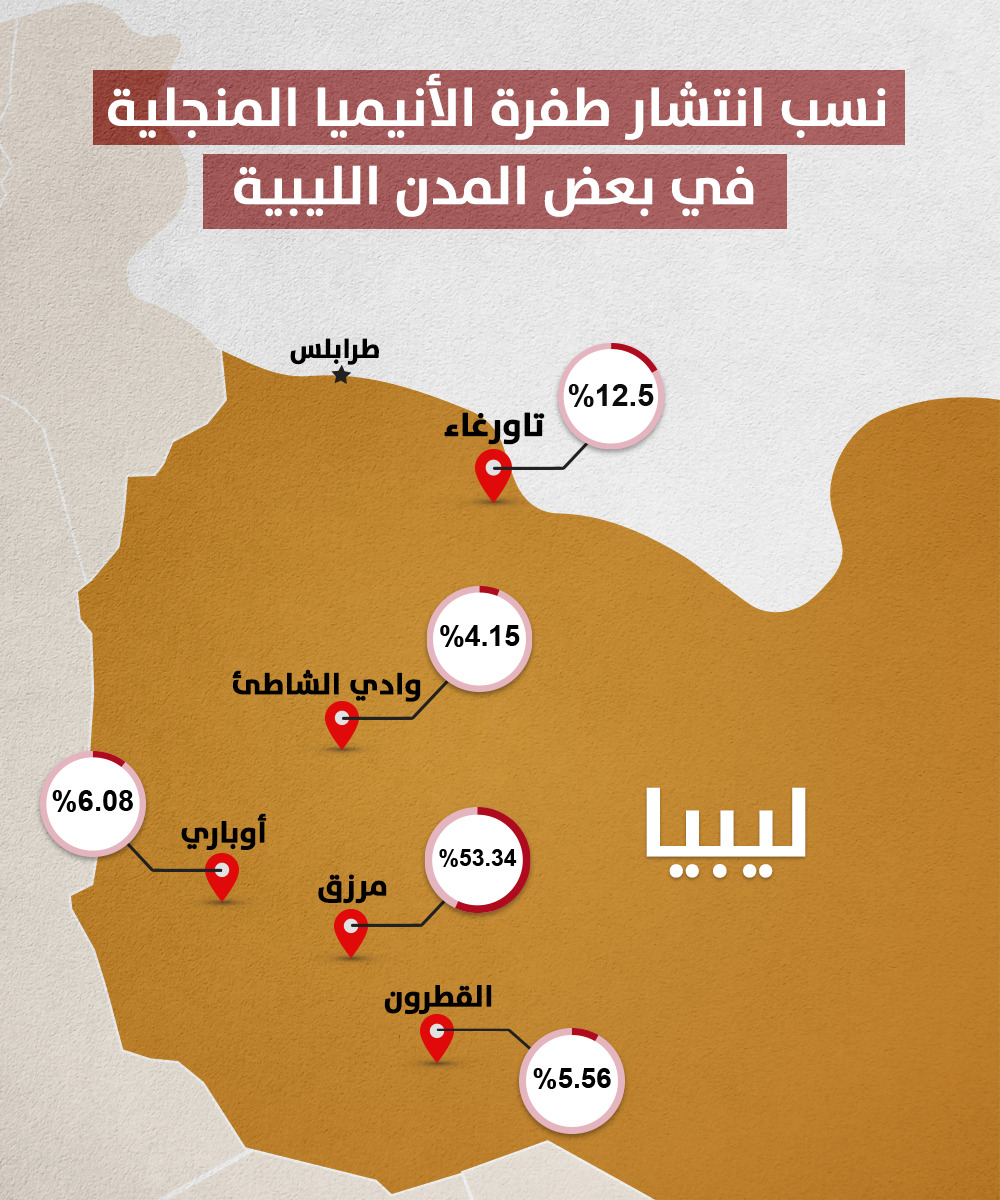
نسبة انتشار الطفرة المنجلية في بعض المدن الليبية بحسب بعض الدراسات العلمية المنشورة [4,5,6,7,8]
{kind=link}
تتراوح نسبة انتشار طفرة الأنيميا المنجلية في مدن الجنوب [4,5,6,7,8] بين 4% إلى 53% أما في تاورغاء فهي تبلغ 12.5%، وبحثًا عن المزيد من التفسيرات التقيت أستاذ علم الوراثة الجزيئية الدكتور محمد مروان في مكتبه بكلية العلوم بجامعة طرابلس، والذي نشر عدة أبحاث علمية حول اضطرابات الدم الوراثية.
عن سبب الانتشار الكبير للمرض في الجنوب وتاورغاء، يقول مروان “هذا يعود إلى أصل طفرة الأنيميا المنجلية، التي يعتقد أنها جاءت كطفرة انتقائية للوقاية من الملاريا، حيث توفر الخلايا المنجلية حماية طبيعية من طفيل الملاريا، كما يعيش الأطفال الحاملون للطفرة المنجلية عند إصابتهم بالملاريا بنسب أعلى من الأطفال الطبيعيين، ما يفسر ازدياد هذه الطفرة في الدول التي ينتشر فيها المرض خاصة في منطقة إفريقيا جنوب الصحراء”.
لهذا يرى أن الأنيميا شائعة بين ذوي البشرة السمراء. “الملاريا كانت متوطنة حتى وقت قريب في الجنوب الليبي، لهذا فإن انتشار الطفرة المنجلية كبير بين الفزازنة (سكان إقليم فزان)، أما مدينة تاورغاء الساحلية فكانت تاريخيًا مركزًا بارزًا لتجارة العبيد”، حسب قوله.
من المثير للاهتمام أن مدينة مرزق في الجنوب بلغت نسبة الإصابة بالمرض فيها 10% فيما تجاوزت نسبة الحاملين للطفرة المنجلية الـ 53%. فبحسب دراسة [5] نشرت عام 2015 أرجع العلماء المشاركون فيها السبب إلى نسبة زواج الأقارب العالية والتي بلغت 72% بين أفراد العائلات العشرين التي خضعت للدراسة، وأيضا إلى كبر حجم العائلات الذي يصل في المتوسط من 6 إلى 10 أطفال لكل عائلة، وهو عامل آخر مهم يزيد من تردد ظهور سمة الأنيميا المنجلية.
لأن الوقاية خير من العلاج
الدكتور مروان يشدد على ضرورة إجراء المزيد من الدراسات والمسوحات على المستوى الوطني، لمعرفة نسب انتشار هذا المرض في المجتمع، مشددًا على وجوب صدور تشريعات تُلزم الأقارب بإجراء فحص جيني قبل الزواج، أسوة بالدول الأخرى التي ترتفع فيها نسب الأمراض الوراثية.
كما كان للحاجي رأي مماثل، حيث رأت أن تقنين زواج الأقارب أمر ضروري في ظل انتشاره في المجتمع الليبي بمعدلات كبيرة، خاصة وأن الأمر لا يرتبط بالأنيميا المنجلية فحسب، بل بعدة أمراض وراثية أخرى بعضها قد يكون قاتلًا، مؤكدة أن نشر الوعي في المجتمع حول مخاطر زواج الأقارب له دور كبير في تجنيب الأسر تكرار مثل هذه المآسي.
العلاج الجيني.. رجاء في المستقبل
إلى الآن لا يوجد علاجٌ شافٍ من مرض الأنيميا المنجلية، والمصاب به لا يعيش لأكثر من أربعة أو خمسة عقود على الأكثر، والعلاج الوحيد المتاح حاليًا هو زرع نخاع العظام، ونظرًا لأن عمليات الزرع يمكن أن تكون لها آثار جانبية خطيرة، فعادة ما يتم استخدامها فقط في الأطفال المصابين بفقر الدم المنجلي الشديد. ولكي تنجح العملية، يجب أن يكون نخاع العظم متطابقًا تمامًا، وعادةً ما يكون أفضل متبرع هو الأخ أو الأخت [2]، فزراعة نخاع من متبرع خارجي قد تزيد احتمالية رفض الجسد للنخاع الجديد.
لكن في فبراير/ شباط الماضي نشرت دورية نيو إنجلاند الطبية [9] نتائج المرحلتين الأولى والثانية من تجارب سريرية أجريت في الولايات المتحدة الأمريكية على علاج جيني من جرعة واحدة اسمه لينتي جلوبين قادر على تصحيح شكل خلايا الدم المنجلية، ومنع نوبات الألم الشديدة من الحدوث.
قام العلماء باستخلاص الخلايا الجذعية المسؤولة عن تصنيع كريات الدم الحمراء من دماء المرضى، وتعديلها وراثيًا بإدخال نسخة سليمة جين من جلوبين – بيتا إليها، ثم قتل الخلايا المتبقية في النخاع بواسطة علاج كيميائي لإفساح المجال للخلايا المعدلة وراثيًا التي يتم حقنها فيما بعد لتتخذ موضعًا في نخاع العظم وتبدأ في إنتاج كرات دم حمراء جديدة معافاة خالية من الطفرة المنجلية.
وتم متابعة المرضى بعد الحقن لمدة 3 سنوات، وكانت النتائج مبهرة للغاية في منعها لأي نوبات ألم، وصار المرضى يعيشون حياتهم بشكل طبيعي تمامًا [10].
أسامة اليوم طالب بالصف الأول الثانوي يحاول أن يعيش حياة طبيعية مثل سائر أقرانه، رغم سياج الإجراءات الوقائية المحكم الذي يحيط به؛ فهو يتلقى تعليمه في مدرسة خاصة، داخل فصل دراسي مكيف، ويتجنب الحضور عندما يمرض أي من زملائه حتى لا تنتقل له العدوى. كما يتحاشى الإرهاق البدني ويحرص على شرب المياه بكثرة طيلة اليوم. لكن كل هذا الاحتراس لم يمنع أن يمر كل فترة وأخرى بنوبة من الألم يدخل على إثرها المستشفى.
لكن الدكتور هويدا الحاجي تُبدي تفاؤلها من ثورة العلاج الجيني القادمة، التي قد تمثل رجاءً لحالات شبيهة بأسامة “هناك عدة دراسات تجرى في العالم على العلاج الجيني للأنيميا المنجلية، وأتوقع أنه خلال 5 أو 6 سنوات على الأكثر سنكون على موعد مع أول علاج جيني يمنح تصريحًا بالاستخدام مع المرضى”.
{kind=link}
المصادر:
1. Sickle Cell Disease (Genes & Disease)
2. Complications and Treatments of Sickle Cell Disease
4. The Incidence of Hemoglobin S in Taourga Region, Libya
5. Prevalence of HbS Gene in Marzouk Region of Southern Libya
6. Prevalence of hemoglobin S in Wadi Eshati Region – South Libya
7. دراسة مبدئية عن مدى انتشار خضاب الدم المنجلي في المترددين على مستشفى القطرون العام.
8. دراسة مبدئية عن مدى انتشار خضاب الدم المنجلي في المترددين على مستشفى أوباري العام.
9. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease
10. Experimental Gene Therapy Reverses Sickle Cell Disease for Years
The post الأنيميا المنجلية: لعنة زواج الأقارب في ليبيا appeared first on عين ليبيا | آخر أخبار ليبيا.